OIST 博士課程学生のタイムライン
OISTの博士課程に所属して3年目になったので、これまでの流れをまとめてみます。
目次
OISTに入学する前の準備などはこちらの記事に書いています。
-
主な登場人物
- メインの指導教員 (Thesis Supervisor): ローテーション終了後から4年間所属する研究室のPIの先生を指します。
- メンター (Academic mentor): 在学中に何でもアドバイスをしてもらえる先生です。
- ローテーションの指導教員: ローテーション期間中にお世話になる先生です。
- Co-supervisor またはThird Committee Member: あなたの研究についてアドバイスをくれる3人目の先生です。(メインの指導教員、メンターに次ぐ3人目という意味)
- Proposal の examiner (OIST 内部の先生 / 外部の先生): 2年次にproposalを審査していただく先生です。
- メインの指導教員 (Thesis Supervisor): ローテーション終了後から4年間所属する研究室のPIの先生を指します。
1年生
ローテーションと授業をこなします。
- 入学直後に、メンターの先生を誰にするかと、在学中に取る授業を決めます。メンターの先生を誰にするかについては、学生課から自分と分野の近い先生が紹介されるかと思います。後から変更することも可能です。
授業決めはメンターとしっかり相談してもいいし、特に相談しない人もいるようです。(制度上メールでメンターの了承を取る必要があります)
授業も後から変更できます。 - メンターとのmeeting(最低でも年に1回。3年次からはResearch Progress Reviewに切り替わる)
定期的にmeetingしてもらいます。メンターも時間がないので、要点をまとめて話をすると良いでしょう。meetingが終わったら、メンターの先生から学生課にレポートを提出していただきます。 - Rotation proposal と Rotation report
ローテーションプロポーザルではA4 1ページでこれからやることの提案書を書きます。各ローテーションが始まって1ヶ月が締め切りです。
ローテーションレポートはA4 3ページでローテーションでやったことをまとめたレポートを書きます。ローテーションが終わる月の最終日が締め切りです。分量は短いですが、練習だと思ってできるだけ良いものを作りましょう。 - おすすめの1年生の過ごし方
(2年生は結構忙しくなる人が多いので) 1年生のうちに予備的な研究成果を出したり、文献調査をたくさんしておくのが良いかと思います。また、運要素もありますが学振の業績欄に書くことを見越して、論文を書いて学術誌に投稿することにトライしてみましょう。
手法としては、
・メイン所属ラボで1年くらいで論文化できそうな小さいテーマに取り組む
・ローテーション先のラボで既に論文化しそうなプロジェクトを手伝って共著に入れてもらう
・OIST入学前の所属大学での卒論や修論を論文化する
等です。
いずれにしても早めに相談に行くにこしたことはないです
ちなみに、自分のメインラボをA研究室としたとき、ローテーションでB研究室に所属している際にも、空いた時間でA研究室に出入りして実験したりすることは可能です。(もちろん両ラボの先生に事前に許可を取っておく)
2年生
2年生での大きな目標はResearch proposal の提出 & Proposal exam の突破です。
単位が取りきれてない場合は授業も取りましょう。
ほとんどの学生は学振も書きます。
OIST学生はもともと大学から給料が支払われていますが、学振を取るとさらに給料が数万アップ + 特別研究員としての研究費を獲得することができます。
PCDという授業の一貫でグループプロジェクトというのもあります。
- PCD グループプロジェクト
入学年度の1月くらいに同学年の学生と3-5人(?) のグループを作ります。グループになったメンバーと協力し、数ヶ月かけて何か地域貢献系のプロジェクトをやります。僕は沖縄の高校生に向けた科学教室を開催しました。このカリキュラムはコロナウイルスの感染状況にかなり影響を受けるため、今後どのような形で行われるのかは不明です。 - Provisional thesis proposal title & scope (入学してから16ヶ月が締切)
4ヶ月後に締め切りが控えるプロポーザルの暫定タイトルとアブストラクトを書きます(実際に提出するプロポーザルは、ここから内容が変わっても問題ないです)。この書類には特に評価がつきませんが、プロポーザルの下準備になるのでしっかり書きましょう。 - 論文指導委員 (Thesis Committee) の決定
論文指導委員は、自分のPI+2人の先生からなる計3人の指導チームのことで、海外のPhD課程ではよくある制度のようです。今後ともこの3名の先生方にはお世話になることになります。
具体的には、
・メインの指導教員
・メンター
・Co-supervisor または Third Comittee Member
の3名の先生を指名します。この3名は、プロポーザルの審査員を指名するまでに決定し、承認して頂く必要があります。
Co-supervisor または 3rd committee memberについて。
あなたの研究についてアドバイスをくれる3人目の先生です。
異なる2つの研究室の要素を取り入れた研究を行いたい場合、メインの指導教員に加えて、さらにもう1人の先生にCo-supervisor (指導教員その2) になってもらうことができます。その場合は3rd committee memberとして誰かについてもらうことはなくなります。メインの指導教員は定年前でなければいけませんが、Co-supervisor や 3rd commitee memberに指名する先生には年齢制限がありません。 -
Thesis proposal (入学してから20ヶ月が締切)
在学中に行う研究について、研究計画書を提出します。プロジェクトの背景の包括的なレビュー、研究のゴール、3~5つの到達目標、それぞれの目標を達成するためのアプローチ、既に得ている予備的な結果、卒業までのタイムライン等で構成されています。分量に特に規定はないですが、20 ~ 数十ページくらいの書類になる人が多いと思います。
締切の前に(理想は1ヶ月ほど前)、論文指導委員の先生方にproposalの完成前原稿を提出し、チェックを受けてサインをもらいます。プロポーザルの提出には論文指導委員の先生方全員からサインをもらう必要があります。 -
プロポーザルの提出前後で、プロポーザルの審査をしていただく審査員の先生を決めます。
OIST内部の先生から1人、外部の先生から1人指名します。
外部の先生がメインの審査員になります。世界中の先生の中から、審査をしていただきたい先生に依頼をします。どの大学の先生でも基本的に大丈夫なはずですが、最低一人はPhD学生を卒業させている必要があります。この審査会をきっかけにコネクションが生まれたりします。
(指導教員から、指導教員の知り合いの先生を外部審査員に指名するように勧められるケースもあります)
ボランティアで審査をしていただく先生を探すことになるので、なかなか先生が見つからず、審査会の日程が後ろ倒しになる場合もあるようです。 -
Thesis proposal Examination (プロポーザル審査会) (入学してから24ヶ月が締切)
Thesis proposalに書いた内容に基づき、内部審査員、外部審査員、チェアの3名に向けてチョークトークを行います。(実際にはホワイトボードです。)
審査会に持ち込んで良いのは事前に提出したThesis proposal (と持ち込みたければ使い慣れたホワイトボードマーカー) のみで、パソコン等の持ち込みはできません。
審査会の日取りは、審査員の先生方のスケジュールに依存します。 -
実際に行ったプロポーザル審査会対策
僕の場合、審査会の日程が決まった時点で、実験は片手間でできるクローニングや細胞のお世話に切り替えました。
朝決めた時間に来て論文 (特に審査員の先生が過去に出版した論文) を読み、お昼過ぎの少し眠くなる時間帯に実験をしながら目を覚まし、夕食前後はセミナールームでチョークトークの練習をする、という生活をおくりました。ラボのみなさんを集めて練習会に付き合っていただいたりもしました。友人や3rd committeeの先生のラボの方にも発表を見てもらいました。(見ていただいた皆様、本当にありがとうございました)
zoomで自分の発表を録画して練習する方もいるようです。Examの準備に際して参考になったwebサイト
www.chem-station.comほとんどの場合、すぐに落ちて退学になることはなく、Pass (一発合格), minar revision (軽い修正), major revision (大幅な修正) のいずれかの結果が来るはずなので、それぞれの案内に従います。
-
学振を書く
OISTのカリキュラムに含まれてはいませんが、OIST学生も学振に申請することが強く推奨されています。内部で先輩採択者を交えた説明会があったり、学生課で採択された先輩の申請書を閲覧できたりします。
ちなみに、既に色んな人が書いていることですが、学振を書くにあたって個人的に大事だと思ったことをまとめた記事はこちらです。
入学時期によってプロポーザルを書くタイミングと学振を書くタイミングが被ったりします。
学振の学内締め切り:5月末
プロポーザルの締め切り
5月入学の人 → 書類:12月末、審査会:4月末
9月入学の人 → 書類:4月末、審査会:8月末
1月入学の人 → 書類:8月末、審査会:12月末
3年生から5年生
バリバリ研究を進めて論文を書きます。
外部の研究費にもじゃんじゃん申請します。
授業も取りたいのがあれば取ります。
(特にキャリア形成系のセミナーの案内はよく来ます)
1年に一度、論文指導委員 (Thesis Committee) の先生方とmeeting (Research Progress Review) を行います。
最後に博論を書いて審査会でプレゼンを行います。
- 年に一度の進捗報告会 (Research Progress Review)
1年間の進捗状況と今後の計画を発表して、論文指導委員の先生方(自分のPI+2人の先生からなる計3人の指導チーム)からアドバイスをもらう会です。学会発表の練習だと思って頑張りましょう。
-
論文指導委員の先生方とのmeetingについては次の記事に良くまとまっていますので一部引用させていただきます。(引用文中のTACというのがResearch Progress Reviewに対応しています。)
(引用始め)
TACは、研究者としての能力を審査される、というよりは自分のプロジェクトについて外部の専門家のディスカッションし、意見を貰う機会と認識しています。
TACでは、発表や質疑応答に対する評価やフィードバックだけでなく、プロジェクトの方向性、取捨選択、ストーリ構成、研究の意義、研究結果を論文にまとめるタイミングなど様々なアドバイスを複数の専門家の視点からもらえます。例えば、私の場合は、イギリスの大学や別の研究科の教授を含めた4人がTACメンバーで、1時間半〜2時間くらいの時間をかけて、次にどんな実験やったら面白そうか、とか、この研究室とコラボレーションしたらどうか、とか、この実験をするにはこの点に注意しないといけない、といった意見をいただきました。
TACメンバーの研究が、基礎か応用か、動物をモデルとして用いるか植物か、ドライな研究かウェットか、どこの国で研究をしているかによって、プロジェクトの方向性や売り込み方、ストーリ構成、考え方が少しづつ違っていて、幅広い意見をもらえるため、大変勉強になります。
また、自分の研究が一般的にどう評価されるかを知ることができ、何よりも、経験も知識も豊富な専門家たちに時間をいただいて、自分のプロジェクトについてディスカッションできるのはとても貴重な機会です。また、PIの指導方法等に問題がないかどうかもこのTACで相談することができ、問題が発生した場合は、TACメンバーが問題解決に働いてくれます。
(引用終わり) - 以下はOIST学生が実際に行う流れです。
meetingの締め切り日の1ヶ月以上前に、学生側から各先生に日程調整のメールを出します。日程が決定したら各先生に確認の連絡をし、学生課のweb formからも申請を行います。
要旨、イントロ、これまでに得た結果、今後のタイムラインを含めた数ページのレポートを書きます。これはmeetingが終わったら締め切りまでに学生課に提出することになります。
ほとんどの場合、発表にはパワポを使うので、それの準備もします。
meetingの10日以上前には、レポートとスライドをメインの指導教員に添削してもらうと良いと思います。
meetingの前日くらいになったら、リマインドも兼ねて各先生にレポートを送っておきます。
当日は発表をし、フィードバックをいただきます。
終了後に先生方には評価書を書いていただくので、評価書はここから提出して下さいね~ (提出先のlinkを添えて) のメールを送ります。
最後に、各先生に送ったレポートを学生課のweb formにも提出して終了です。
-
- 5年目の最後までに博論を書き、博論審査会をして合格したら卒業です。
以上5年間をまとめるとこんな感じです (OIST公式より引用)。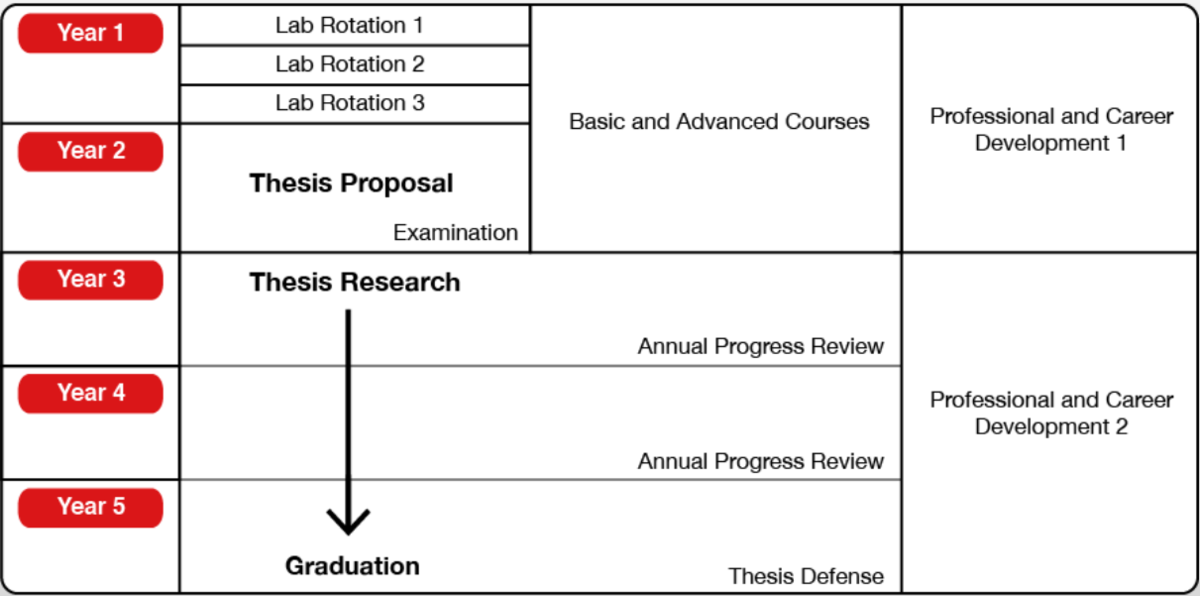
公式に載っていなさそうなQ&A
- OISTに入学したら最初の学振 (DC1) はいつ出す?
2年生の3月~4月に準備を始め、学内締め切りの5月末 - 6月頭に提出します。
5月, 9月, 1月のどの時期に入学していても同じです。
(これは、DC1の応募資格が、採用開始時の在学月数によって決まるからです。採用開始時に "一貫制の博士課程第3年次相当(在学月数24 ヶ月以上36 ヶ月未満)に在学する者" という規定です。募集要項(PD・DC2・DC1) | 特別研究員|日本学術振興会) - OISTで学生をしながら応募できる外部資金は?
次年度の4月1日時点でOISTの学生(かつ35歳未満)の場合、任意のタイミングで(入学して初年度でもいけるはず)、笹川研究助成に出すことが出来ます。締め切りは10月ごろ?
僕の専門に寄っててあれですが、他には以下のようなものがあります。これ以外にもあったら教えて下さい。
孫正義財団 (25歳以下)
戸部眞紀財団 (30歳以下)
ANRI (年齢制限無し)
日本ジェネティクス (生物系、35歳以下)
リカケンホールディングス (生物系、30歳以下)
ちなみにJST次世代や文科フェ(参考:こばしり on Twitter)はOISTの学生は対象外なので出せません。 - 締切に間に合わないとどうなる?
基本的にどんな締切も延長申請をすることで延長することが可能なはずです。ただし、就学年数は5年と決められており、それを過ぎると経済支援は打ち切られてしまうようです。 - 卒業要件は?
OISTとしては、論文の投稿まですれば卒業できるという話を聞いています (年度によって制度が改訂されるので要問合せ) 。また、卒業要件に独自の基準を設けている研究室もあるようです (最低2本は論文出してから卒業しようね~的な) 。
OISTは、学部卒で来た人も修士卒の人もほぼ同じ扱いを受けます (大きな違いは修士卒だと取らないといけない授業の単位数が少ないくらい) ので、学部卒からOISTに入学した人も、修士卒の人も、博士号にふさわしいと判断されれば3年くらいで卒業することは理論上可能だと思います。
【学振対策】差がつきやすいポイントと学部生からでもできる準備
本記事では、学振DC申請書を見ていて内容に差がついたと思われるポイントを3つ挙げて分析し、差をつけるための準備について考察しています。
差がついたポイント
1. 研究計画の完成度、申請者の貢献度
研究計画の完成度は一番差が出ると思います。
学振や科研費の申請書は、特に「1ページ目」で全てが決まると言われています。
ⅰ. 最初に気にすべきなのは先行研究との差別化がクリアになっているかどうかです。上手い人の申請書は、先行研究と比較したときの研究計画の位置づけ、どこに独自性があるのかが分かりやすく書かれています。
例えば、分かっていない(or やられていない)ことがあります→だから私がやります、としか書かれていない研究計画はダメな例です。
読んでいてしっくりきやすいのは、
・重要な課題があります(分野外の人にも分かるように強調する)
↓
・その課題についてはxxxのようなアプローチが取られてきました
↓
・そこに新たな視点を持ち込みます
↓
・分かっていなかった重要なことが分かります
という流れです。
ⅱ. 申請者自身が主体的に研究を進めているように見えるかどうかにも差がありました。ラボの強みが書かれているだけなのは低い評価になってしまうと思います。
【良い研究計画を書くための準備】
- 綿密なフィードバックをもらえる環境づくり
=グラント書くのが上手い人(過去の採択者やポスドク以上の科研費採択経験者)から綿密なフィードバックを受けられる環境に身を置く。
申請書を良いものにするのに万能な方法などなく、ひたすらフィードバックをもらいながら、地道に改訂を繰り返していくしかないと思います。なので、そのようなフィードバックを受けられる場所に身を置くことが最重要だと考えます。
ちなみに、大学院生くらいで文章書くときにおすすめの考え方が、苦しみながら遠回りして書いた方が申請書を書く力がつく、というものです。学振を書く過程で、文章作成能力が鍛えられるので、最初から完璧なものを書けると思わないほうが良いです。構成の大幅な変更やほぼ全部書き直しをくらっても、それまでの時間は無駄ではありません。学振は教育コンテンツだという心持ちで根気強くやりましょう。- 発展性、インパクトや意義が大きいテーマに着手できるラボを選ぶ。(学振採択常連ラボに入ってラボから強いテーマもらって学振取るのもひとつの戦略です)
- 学振に集中できるようなラボを選び、準備は早めに始める
提出前の2-3週間前までに予備データの取得や論文出版が完了していて、先生から頼み事をされることも少なく、学振申請書の大幅な改訂に耐える時間を作れるのがベスト。理想のタイムラインは、1ヶ月以上前に書き始めており、3-4週前に第一稿が書き終わっていること。
2. 予備データ、業績
予備データがあるかないかでも差がでます。これまでだれもやってこなかった何かしらを既に明らかにしていることで、申請者の研究遂行能力が推察できますし、研究計画の実行可能性も高そうだな、となります。
また、「着想に至った経緯」の部分で、申請者自身の予備データに基づいて着想したと書けば、ラボの強みだけではなく、申請者自身が計画の立案に貢献していることが伝わってきます。研究の独自性の担保にもなります。
また、当然ですが業績があった方が有利です。
【 業績、強い予備データをつむための準備】
- 論文が書けそうなら何としてでも書く。(ハゲタカじゃなければIF低くてもOK)
そうでないなら、学振で評価され得る業績は意外となんでもありなので頑張る。
(学会・研究系国際大会 (iGEM等)などにおける発表や受賞、奨学金助成金などの採択経験)
評価としては、やはりファーストオーサーの論文を持っていることが最も高評価に繋がりやすいと思います。
個人の頑張りとラボ選びを含めた準備の早さが重要です。運要素あり。
僕の場合は、学部2年からラボに行ってたのが有利に働きました。- (番外編)申請区分を工夫して選ぶ
基本的に論文が出にくい分野の申請区分を選んだほうが有利です。
(業績がそこまで強くない人の中で上位20%を狙う方が勝率が高い)
過去の採択者をチェックしたり、指導教員や先輩に相談するなどして、出せそうな区分の中で一番有利な区分を選びましょう。
ちなみに、申請区分を選ぶのは提出の直前ではなく、申請書を書き始める前が推奨です。申請書の構成を考える段階から申請区分の審査員にウケる内容を狙って書いた方が勝率が上がります。
3. 自己分析パートの独自性、研究との関連性
時間に余裕を持って準備ができている方は、ここではほとんど差がつかないと思います。強いて言うなら2点あります。
ⅰ. 申請者の強みやなりたい研究者像に申請者独自の経験が書かれているか、という点です。独自の経験が書いてあると、本当のことを書いているんだなと思えるので評価が高いです。
ⅱ. そのエピソードによってガンガン研究を進めて論文を書いてくれそうなことが伝わってくるか、という点です。これは、自身の強みが研究の遂行力につながるように書かれているかということです。
例えば、「英語力が高いです」「成績が良いです」としか書いていないのはダメな例です。「英語力が高いから、海外の研究者とも共同研究をやっていけるポテンシャルがあります(または、既にそのような実績があります)」、というところまで掘り下げて書くべきです。
【良い自己分析パートを書くための準備】
- 時間に余裕を持って書く
特に自己分析パートは色んな人から添削を受ければ短期間ですごく良くなることが多いです。添削のサイクルを回す時間が取れれば、独自性があって面白いものが書けると思います。
具体的に経験を書くことと、研究に関連した客観的に評価可能な成果に結びつけるのがコツです。
以上3つの準備を踏まえて、
業績(特にファーストオーサーの論文)と強い予備データを取り、グラント書くのが上手い人(できれば直属の先生)から、丁寧な添削を受けれる環境に身を置くことができれば、きっと学振が取れることと思います!健闘を祈ります!
参考になりそうな文献
学振本は以下のスライドで代用可能
(学振本の中にはさらに過去の採択者の申請書が公開されている)
https://www.slideshare.net/tonets/gakushin23
学振本に加えて、こういう科研費の本も本格的で参考になる
https://www.yodosha.co.jp/yodobook/book/9784758121071/
アポトーシスの直前、小胞体のCa2+量枯渇に依存してCRTが細胞膜表面にさらされる (Cell Death & Differentiation 2007年11月23日号掲載論文)
結論から言うと、アポトーシスの直前、小胞体のCa2+量枯渇に依存してカルレティキュリンという小胞体タンパク質が細胞膜表面にさらされることを示した論文。
本日は「Reduction of endoplasmic reticulum Ca2+ levels favors plasma membrane surface exposure of calreticulin (小胞体Ca2+レベルの低下はカルレティキュリンの膜表面露出を促進する)」という論文で、フランス Centre de Recherche des Cordeliers の Dr. G Kroemer のグループ(どういったラボ?→*1)による研究。(論文サイトへのlink→*2)
構造に分けて解説。
背景 化学物質の投与でがん細胞にアポトーシスを誘導することはがん治療に有益。これまでにアントラサイクリンが細胞死を誘導することが知られていた。
課題 アントラサイクリンによるアポトーシスの前に、カルレティキュリン(CRT)というタンパク質が細胞膜表面に曝されることが知られていたが、その意義に関しては不明である。
目的 CRTの細胞膜へのexposureとCa2+ホメオスタシスの関係を調査する。
方法 神経芽腫細胞株(SH-SY5Y)において、SERCAの阻害やReticulon1Cの過剰発現によりER内腔Ca2+濃度を低下させ、アントラサイクリン投与の影響を見る。
結果① SH-SY5Yは、アントラサイクリン投与でもCRT exposureが誘導されないが、Reticulon1Cの過剰発現と組み合わせるとCRT exposureが起きる。
結果② brefeldin Aにより小胞体からゴルジ複合体へのタンパク質輸送阻害を行うと、CRT exposureがキャンセルされる。
結果③ Reticulon1Cの過剰発現によりER内腔Ca2+濃度が低下している。
結果④ 細胞内(および小胞体)Ca2+のキレート、IP3受容体のリガンド結合部位の発現、およびSERCAポンプの阻害は、SH-SY5YおよびHeLa細胞において、小胞体Ca2+濃度を減少させ、細胞表面でのアポトーシス前CRT曝露を促進した。
解釈 アポトーシス前のCRT曝露は、ERのCa2+枯渇によって誘導される。
最近出た論文では、細胞膜表面のCRTをマクロファージが死細胞の目印として認識しているという報告がある(このブログで以前取り上げました↓)
アポトーシスの前にCRTを出すのは、恐らくアポトーシスの後だと細胞が死んでいるのでCRTを出せないのだろう。しかし、ERのカルシウム量を感知してCRTを提示するという仕組みだと、アポトーシスしていない細胞でもERのカルシウムが枯渇するとCRTを提示してしまうので、他にも制御する仕組みがありそう。
*1:このグループは内的および外的ストレスに対する細胞の応答を、細胞死などの観点から研究しているラボのようです。http://www.kroemerlab.com/index.php/science/researchより。
KClによる刺激で神経のERが断片化 (Journal of neuroscience research 2011年5月2日号掲載論文)
結論から言うと、脳海馬スライスの神経では、KCl刺激によるCa2+流入よって、ERが断片化することを示した論文。
本日は「Potassium-induced structural changes of the endoplasmic reticulum in pyramidal neurons in murine organotypic hippocampal slices. (マウス海馬のスライスにおける錐体ニューロン内ERのKCl刺激による構造変化。)」という論文で、スウェーデン Laboratory for Experimental Brain Research, Department of Clinical Sciences Lund, Lund University の Håkan Toresson のグループ(どういったラボ?→*1)による研究。(論文サイトへのlink→*2)
ちょっと構造に分けて説明していきます。
背景 ERの構造は、細胞内における同化、ストレス応答、シグナル伝達等の調節に重要な役割を果たしている。ERは一般的に連続的な構造をしているが、一時的に構造変化を起こして断片化した状態になることがある。
課題 ERの断片化の発生機序、生物学的意義に関しては詳しく分かっていない。
方法 ERを標的とした蛍光タンパク質を発現させた脳の海馬スライスに対して、過度に神経脱分極を誘導するレベルのKCl刺激(50mM)を加えることで、ERの断片化を誘導し、断片化の機構を調査した。
結果① KCl刺激はERの断片化を誘導する。K+が基底濃度(2.5mM)に戻るとERは融合して元の構造に戻る。この融合・分裂の過程は何度も繰り返し誘導することができる。
結果② Ca2+取り込みチャネルであるNMDA-Rの阻害や細胞外Ca2+の除去でERの断片化はキャンセルされる。
結果③ ERの断片化は35℃よりも30℃で顕著である。
結論 脳海馬スライスの神経では、KCl刺激によるCa2+流入よって、ERが断片化する。
意見 今回のKCl刺激は生理学的な神経の脱分極と比べるとかなり強いため、通常の神経の脱分極ではどの程度ERが断片化するのか疑問である。Ca2+取り込み/放出チャネルを始めとしたER局在タンパク質はもともと均一に分布している訳ではないが、Ca2+に依存したERの断片化はさらなるタンパク質の局所化を引き起こすことになるだろう。どのような分子が、どのようにして可逆的なERの融合と分裂を担うのか、今後の研究で明らかになると面白いと思う。
ER 局在のp53 がCa2+に依存してアポトーシスを制御する (PNAS 2015年1月26日号掲載論文)
結論から言うと、
p53はERとミトコンドリアのコンタクトサイトに局在し、ER へのCa2+取り込みを促進する。Ca2+濃度が高くなったER は、MAM を通してミトコンドリアにCa2+を伝達し、ミトコンドリア内のCa2+が過剰になる。これによりアポトーシスが誘導される。以上の経路の存在が示唆された論文。
本日は「p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner (小胞体のp53はCa2+依存的にアポトーシスを制御する)」という論文で、イタリア Department of Morphology, Surgery and Experimental Medicine, Section of Pathology, Oncology and Experimental Biology, University of Ferrara の Paolo Pinton のグループ(どういったラボ?→*1)による研究。(論文サイトへのlink→*2)
ちょっと構造に分けて説明していきます。
背景 p53はミトコンドリアを介してアポトーシスを誘導することが知られていた。
課題 p53がどのようにしてアポトーシスを誘導するのか、詳細な分子機構の理解は完全ではない。
結果① p53が小胞体とミトコンドリアのコンタクトサイト(MAM; mitochondria-associated membranesの略)に局在する。
結果② p53が活性化するとER内へのCa2+取り込みが促進する。
結果③ ER上のp53はミトコンドリア内Ca2+濃度を上昇させ、アポトーシスを誘導する。
結果④ p53はER上のSARCA(ERにCa2+を取り込むポンプ)に直接結合して、SARCAの酸化還元状態を変化させる。
考察 以上の結果を合わせ、Ca2+濃度が高くなったERは、MAMを通してミトコンドリアにCa2+を伝達し、ミトコンドリア内のCa2+が過剰になる。これによりアポトーシスが誘導される、という経路の存在が示唆された。
結論 p53はERとミトコンドリアのコンタクトサイトに局在し、ERへのCa2+取り込みを促進する。Ca2+濃度が高くなったERは、MAMを通してミトコンドリアにCa2+を伝達し、ミトコンドリア内のCa2+が過剰になる。これによりアポトーシスが誘導される。以上の経路の存在が示唆された。
意見 p53は転写因子としてよく知られているが、核に移行せずともアポトーシス誘導することができるという報告だった。どこまで多機能なのか。
興味を持たれた方はabstractもどうぞ。
The tumor suppressor p53 is a key protein in preventing cell transformation and tumor progression. Activated by a variety of stimuli, p53 regulates cell-cycle arrest and apoptosis. Along with its well-documented transcriptional control over cell-death programs within the nucleus, p53 exerts crucial although still poorly understood functions in the cytoplasm, directly modulating the apoptotic response at the mitochondrial level. Calcium (Ca2+) transfer between the endoplasmic reticulum (ER) and mitochondria represents a critical signal in the induction of apoptosis. However, the mechanism controlling this flux in response to stress stimuli remains largely unknown. Here we show that, in the cytoplasm, WT p53 localizes at the ER and at specialized contact domains between the ER and mitochondria (mitochondria-associated membranes). We demonstrate that, upon stress stimuli, WT p53 accumulates at these sites and modulates Ca2+ homeostasis. Mechanistically, upon activation, WT p53 directly binds to the sarco/ER Ca2+-ATPase (SERCA) pump at the ER, changing its oxidative state and thus leading to an increased Ca2+ load, followed by an enhanced transfer to mitochondria. The consequent mitochondrial Ca2+ overload causes in turn alterations in the morphology of this organelle and induction of apoptosis. Pharmacological inactivation of WT p53 or naturally occurring p53 missense mutants inhibits SERCA pump activity at the ER, leading to a reduction of the Ca2+ signaling from the ER to mitochondria. These findings define a critical nonnuclear function of p53 in regulating Ca2+ signal-dependent apoptosis.
(私訳と勝手な注釈)
腫瘍抑制因子p53は、細胞の癌化や腫瘍の進行を抑制するための重要なタンパク質です。様々な刺激によって活性化されたp53は、細胞周期停止とアポトーシスを制御する。核内での細胞死プログラムを制御する転写制御に加えて、p53は細胞質においても重要な機能を発揮し、ミトコンドリアレベルでのアポトーシス応答を直接調節していますが、その機構はまだ十分に理解されていません。小胞体とミトコンドリア間のカルシウム(Ca2+)移動は、アポトーシスの誘導において重要なシグナルである。しかし、ストレス刺激に応答してこのフラックスを制御するメカニズムはほとんど不明である。本研究では、WT p53が細胞質において、小胞体とミトコンドリアの間の特殊な接触領域(ミトコンドリア関連膜)に局在することを示した。本研究では、WT p53がストレス刺激を受けると、これらの部位に蓄積し、Ca2+の恒常性を調節することを明らかにした。また、WT p53は活性化されると、ERのサルコ/ER Ca2+-ATPase(SERCA)ポンプに直接結合し、その酸化状態を変化させてCa2+負荷を増加させ、その後ミトコンドリアへの移行を促進することを明らかにした。その結果、ミトコンドリアのCa2+負荷が増加し、ミトコンドリアの形態が変化し、アポトーシスが誘導された。WT p53または天然に存在するp53ミスセンス変異体を薬理学的に不活性化すると、ERでのSERCAポンプ活性が阻害され、ERからミトコンドリアへのCa2+シグナル伝達が低下することがわかった。これらの知見は、Ca2+シグナルに依存したアポトーシスの制御において、p53の非核機能が重要な役割を果たしていることを示している。
*1:このグループはCa2+のダイナミクスを分子細胞レベルで研究しているラボのようです。 --The laboratory has developed avouched competences in the studying of calcium (Ca2+) homeostasis, in particular using recombinant proteins such as the Ca2+-sensitive photoprotein aequorins, the mutants of Green Fluorescent Protein (GFP) and different chemical probes. Moreover, we operate as center for cell imaging with particular interest in mitochondrial and Protein Kinase C (PKC) imaging. Finally, the group is involved in the study of adipocytes differentiation of stem cells population derived from different adult tissues. -- http://sm.unife.it/it/ricerca-e-terza-missione/ricerca-1/ambiti/signaltransductionlab/researchより。
DNA 損傷により滑面小胞体が増加, ER-mito 間MCS の形成強化, アポトーシスにつながる (Cell Research 2018年7月20日号掲載論文)
結論から言うと、
DNA 損傷によりp53 が活性化すると、REEP1/2 の発現上昇による滑面小胞体の増加、EI24 の発現上昇によるER-ミトコンドリア間MCS の形成促進が誘導される。その結果ER からミトコンドリアへのCa2+移動が促進されアポトーシスが誘導される、ということを示した論文。
本日は「DNA damage triggers tubular endoplasmic reticulum extension to promote apoptosis by facilitating ER-mitochondria signaling (DNA損傷は滑面小胞体の伸長を誘発し、ER-ミトコンドリアシグナル伝達を促進してアポトーシスを促進する。)」という論文で、中国 Key Laboratory of Cell Proliferation and Differentiation of the Ministry of Education, State Key Laboratory of Membrane Biology, College of Life Sciences, Peking University(北京大学) の Jianguo Chen のグループによる研究。(論文サイトへのlink→*1)
ちょっと構造に分けて説明していきます。
背景 p53はミトコンドリアを介してアポトーシスを誘導することが知られていた。
課題 p53がどのようにしてアポトーシスを誘導するのか、詳細な分子機構の理解は完全ではない。
結果① p53が小胞体とミトコンドリアのコンタクトサイト(MAM; mitochondria-associated membranesの略)に局在する。
結果② p53が活性化するとER内へのCa2+取り込みが促進する。
結果③ ER上のp53はミトコンドリア内Ca2+濃度を上昇させ、アポトーシスを誘導する。
結果④ p53はER上のSARCA(ERにCa2+を取り込むポンプ)に直接結合して、SARCAの酸化還元状態を変化させる。
考察 以上の結果を合わせ、Ca2+濃度が高くなったERは、MAMを通してミトコンドリアにCa2+を伝達し、ミトコンドリア内のCa2+が過剰になる。これによりアポトーシスが誘導される、という経路の存在が示唆された。
結論 p53はERとミトコンドリアのコンタクトサイトに局在し、ERへのCa2+取り込みを促進する。Ca2+濃度が高くなったERは、MAMを通してミトコンドリアにCa2+を伝達し、ミトコンドリア内のCa2+が過剰になる。これによりアポトーシスが誘導される。以上の経路の存在が示唆された。
意見 p53は転写因子としてよく知られているが、核に移行せずともアポトーシス誘導することができるという報告だった。どこまで多機能なのか。
翻訳と無関係にmRNAがERに局在化する (Plos Biology 2012年5月29日号掲載論文)
結論から言うと、mRNA がリボソームに依存せずにER 局在化する経路が存在し、このプロセスにはp180 というタンパク質が必要であることを示した論文。
本日は「p180 Promotes the Ribosome-Independent Localization of a Subset of mRNA to the Endoplasmic Reticulum (p180はリボソームに依存しない小胞体へのmRNAの局在化を促進する)」という論文で、カナダ Department of Biochemistry, University of Toronto の Andrea Daga のグループ(どういったラボ?→*1)による研究。(論文サイトへのlink→*2)
ちょっと構造に分けて説明していきます。
背景 分泌タンパク質や膜結合タンパク質をコードするmRNAの大部分が小胞体(ER)表面(内腔ではないことに注意)で翻訳されている。これらのmRNAの小胞体表面へのターゲティングはシグナル配列によって行われ、その維持はリボソームとトランスロコンの相互作用によって行われる。一方で近年、さらなる別の経路(以下alternative pathway)でmRNAが小胞体に局在化することが明らかになってきている。
課題 alternative pathwayがどのような機構でmRNAをERに局在化するのか不明である。
目的 alternative pathwayに関与するタンパク質を同定し、その機構を解明する。
方法 ER画分からラフER(ポリソームをよく含む)画分を分離し、そのラフER画分にRNAse A処理をしたとき上清に来るタンパク質の質量分析を行う。「ラフERに存在しながら、インタクトなRNAが存在しないと同じ画分に落ちてこないタンパク質群」=「ラフER においてリボソームと独立してRNAに結合するタンパク質」を含む、という仮説にもとづく。
結果① リボソームや翻訳とは無関係にERに標的化され、ERに局在したままであるmRNAが存在する。
結果② 質量分析の結果、このプロセスに関与しているであろうmRNA結合タンパク質; p180を同定した。(p180はもともとERの膜タンパク質として知られていた。リボソームとも相互作用する)
結果③ p180にはリジンに富んだ領域があり、RNAと直接相互作用できることをin vitroで示した。
結果④ p180のKDにより、p180がバルクmRNAのpoly Aの効率的なERへのアンカーリングに必要である(poly Aを標識したFISHでERとの共局在を見ただけなので、poly Aとp180が結合しているかは不明)ことと、placental alkaline phosphataseやカルレティキュリンなどの特定のmRNAのERへのアンカーリングに必要であることを示した
考察 このalternative pathwayは、1)タンパク質選別の正確性を高める、2)mRNAをERの様々なサブドメインに局在させる、という意義があるかもしれない。
結論 mRNAがリボソームに依存せずにER局在化するalternative pathwayが存在し、このプロセスにはp180が必要である。
意見 リボソームが来る前にmRNAをERに局在させることができれば、局所的な翻訳効率が上がりそうだが、そのようなプロセスが必要な生理的条件があるのか疑問が残った。


そのmRNAは、何のためにERを目指すのだろう?
興味を持たれた方はabstractもどうぞ。
In metazoans, the majority of mRNAs coding for secreted and membrane-bound proteins are translated on the surface of the endoplasmic reticulum (ER). Although the targeting of these transcripts to the surface of the ER can be mediated by the translation of a signal sequence and their maintenance is mediated by interactions between the ribosome and the translocon, it is becoming increasingly clear that additional ER-localization pathways exist. Here we demonstrate that many of these mRNAs can be targeted to, and remain associated with, the ER independently of ribosomes and translation. Using a mass spectrometry analysis of proteins that associate with ER-bound polysomes, we identified putative mRNA receptors that may mediate this alternative mechanism, including p180, an abundant, positively charged membrane-bound protein. We demonstrate that p180 over-expression can enhance the association of generic mRNAs with the ER. We then show that p180 contains a lysine-rich region that can directly interact with RNA in vitro. Finally, we demonstrate that p180 is required for the efficient ER-anchoring of bulk poly(A) and of certain transcripts, such as placental alkaline phosphatase and calreticulin, to the ER. In summary, we provide, to our knowledge, the first mechanistic details for an alternative pathway to target and maintain mRNA at the ER. It is likely that this alternative pathway not only enhances the fidelity of protein sorting, but also localizes mRNAs to various subdomains of the ER and thus contributes to cellular organization.
(私訳と勝手な注釈)
分泌タンパク質や膜結合タンパク質をコードするmRNAの大部分が小胞体(ER)表面で翻訳されている。これらのmRNAの小胞体表面へのターゲティングはシグナル配列によって行われ、その維持はリボソームとトランスロコンの相互作用によって行われる。一方で近年さらなる小胞体局在化経路が存在することが次第に明らかになってきている。ここでは、こういったmRNAの多くがリボソームや翻訳とは無関係にERに標的化され、ERに関連したままであることを示している。ERと結合したポリソームと結合するタンパク質の質量分析を用いて、このプロセスに関与しているであろうmRNA受容体を同定した。その中にはp180が含まれており、p180は正電荷を帯びている発現量の高いタンパク質である。我々は、p180の過剰発現が一般的なmRNAとERとの関連性を高めることを示した。さらに、p180には、RNAと直接相互作用することができるリジンに富んだ領域が含まれていることをin vitroで示した。最後に、p180がバルクポリ(A)の効率的なERへのアンカーリングと、placental alkaline phosphataseやカルレティキュリンなどの特定の転写物のERへのアンカーリングに必要であることを示した。以上のことから、我々は、mRNAを標的とし、ERで維持する経路の中でもnon-cannnonicalな経路のメカニズムの詳細を初めて明らかにした。このnon-cannnonicalな経路は、タンパク質選別の正確性を高めるだけでなく、mRNAをERの様々なサブドメインに局在させ、細胞の組織化に貢献している可能性がある。
*1:このグループはmRNAに主眼をおいていて、mRNAの核外への輸送, 適切なmRNAの局在, ERにおける翻訳と細胞質における翻訳の違い、等を研究しているラボのようです。 --1) How are mRNAs exported from the nucleus? 2) How are mRNAs localized to their proper subcellular destination, such as the endoplasmic reticulum? 3) How does mRNA translation in the cytosol differ from translation on the endoplasmic reticulum? -- http://biochemistry.utoronto.ca/person/alexander-f-palazzo/より。このトロント大学のbiochemistryのwebページがカッコよい!
*2:https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.1001336