本日は「VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease (VDACオリゴマーはミトコンドリアの孔を形成してmtDNA断片を放出し、ループス様疾患を促進する)」という論文で、米国 NHLBI (NIHが母体らしい) の Laboratory of Obesity and Aging Research, Cardiovascular Branch の Dr. Jay H. Chung のグループによる研究。(論文サイトへのlink→*1)
Mitochondrial stress releases mitochondrial DNA (mtDNA) into the cytosol, thereby triggering the type Ι interferon (IFN) response. Mitochondrial outer membrane permeabilization, which is required for mtDNA release, has been extensively studied in apoptotic cells, but little is known about its role in live cells. We found that oxidatively stressed mitochondria release short mtDNA fragments via pores formed by the voltage-dependent anion channel (VDAC) oligomers in the mitochondrial outer membrane. Furthermore, the positively charged residues in the N-terminal domain of VDAC1 interact with mtDNA, promoting VDAC1 oligomerization. The VDAC oligomerization inhibitor VBIT-4 decreases mtDNA release, IFN signaling, neutrophil extracellular traps, and disease severity in a mouse model of systemic lupus erythematosus. Thus, inhibiting VDAC oligomerization is a potential therapeutic approach for diseases associated with mtDNA release.
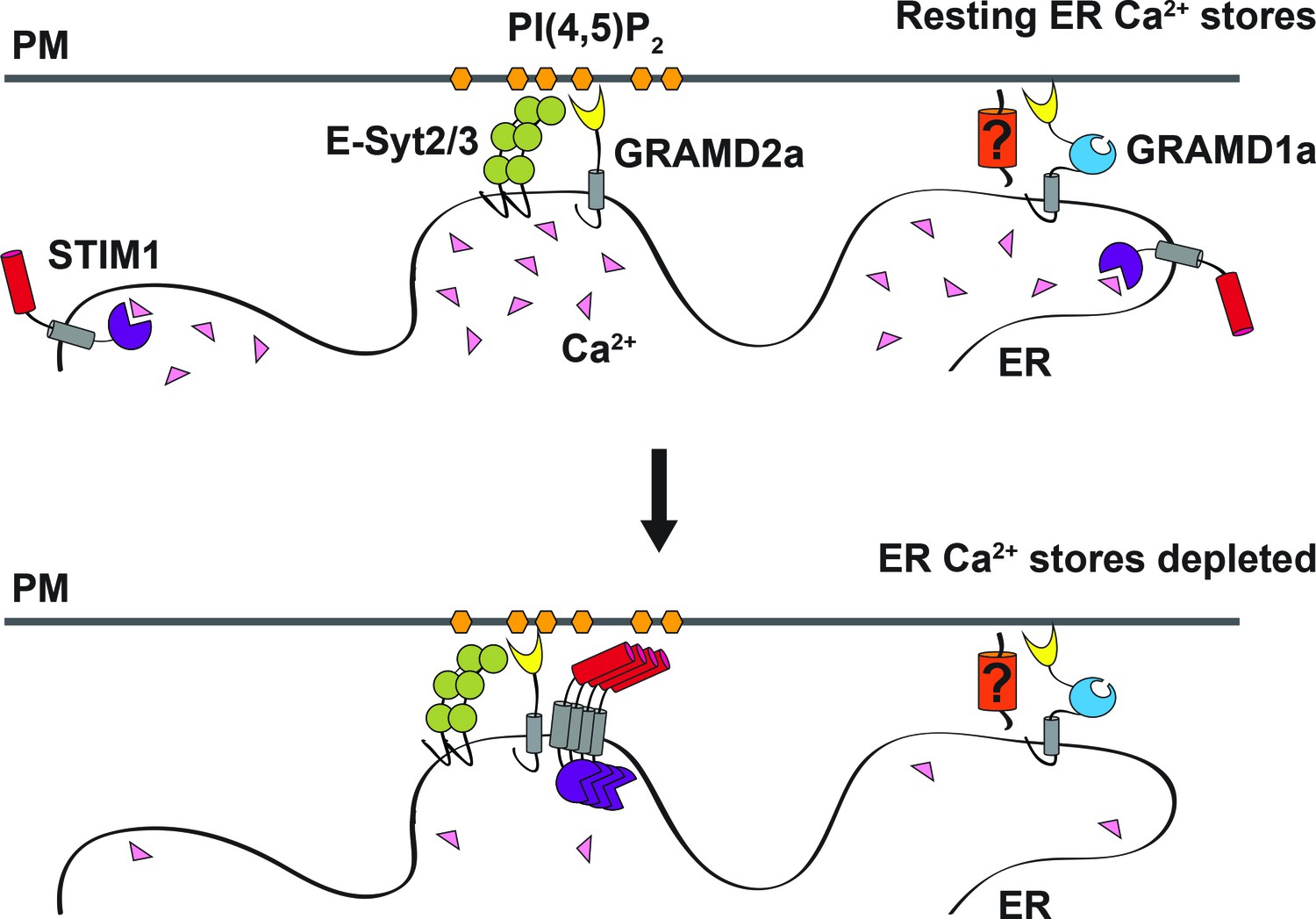
本日は「GRAM domain proteins specialize functionally distinct ER-PM contact sites in human cells (GRAMドメインタンパク質は、ヒト細胞において、ER-PM接触部位を機能的に異なるものに特化させる)」という論文で、米国 カリフォルニア Department of Molecular and Cellular Biology, University of California, Davis の Dr. Jodi Nunnari のグループによる研究。(論文サイトへのlink→*1)
Endoplasmic reticulum (ER) membrane contact sites (MCSs) are crucial regulatory hubs in cells, playing roles in signaling, organelle dynamics, and ion and lipid homeostasis. Previous work demonstrated that the highly conserved yeast Ltc/Lam sterol transporters localize and function at ER MCSs. Our analysis of the human family members, GRAMD1a and GRAMD2a, demonstrates that they are ER-PM MCS proteins, which mark separate regions of the plasma membrane (PM) and perform distinct functions in vivo. GRAMD2a, but not GRAMD1a, co-localizes with the E-Syt2/3 tethers at ER-PM contacts in a PIP lipid-dependent manner and pre-marks the subset of PI(4,5)P2-enriched ER-PM MCSs utilized for STIM1 recruitment. Data from an analysis of cells lacking GRAMD2a suggest that it is an organizer of ER-PM MCSs with pleiotropic functions including calcium homeostasis. Thus, our data demonstrate the existence of multiple ER-PM domains in human cells that are functionally specialized by GRAM-domain containing proteins.
本日は「Mitochondrial calcium overload is a key determinant in heart failure (ミトコンドリアのカルシウム過負荷は心不全の決定的な要因である)」という論文で、米国 ニューヨーク Department of Physiology and Cellular Biophysics, College of Physicians & Surgeons, Columbia University Medical Center の Dr. Gaetano Santulli のグループによる研究。(論文サイトへのlink→*1)
Calcium (Ca2+) released from the sarcoplasmic reticulum (SR) is crucial for excitation–contraction (E–C) coupling. Mitochondria, the major source of energy, in the form of ATP, required for cardiac contractility, are closely interconnected with the SR, and Ca2+ is essential for optimal function of these organelles. However, Ca2+ accumulation can impair mitochondrial function, leading to reduced ATP production and increased release of reactive oxygen species (ROS). Oxidative stress contributes to heart failure (HF), but whether mitochondrial Ca2+ plays a mechanistic role in HF remains unresolved. Here, we show for the first time, to our knowledge, that diastolic SR Ca2+ leak causes mitochondrial Ca2+ overload and dysfunction in a murine model of postmyocardial infarction HF. There are two forms of Ca2+ release channels on cardiac SR: type 2 ryanodine receptors (RyR2s) and type 2 inositol 1,4,5-trisphosphate receptors (IP3R2s). Using murine models harboring RyR2 mutations that either cause or inhibit SR Ca2+ leak, we found that leaky RyR2 channels result in mitochondrial Ca2+ overload, dysmorphology, and malfunction. In contrast, cardiac-specific deletion of IP3R2 had no major effect on mitochondrial fitness in HF. Moreover, genetic enhancement of mitochondrial antioxidant activity improved mitochondrial function and reduced posttranslational modifications of RyR2 macromolecular complex. Our data demonstrate that leaky RyR2, but not IP3R2, channels cause mitochondrial Ca2+ overload and dysfunction in HF.
(私訳と勝手な注釈)
筋小胞体(SR)から放出されるカルシウム(Ca2 +)は、興奮収縮(excitation–contraction: E–C)カップリングに重要です。心臓の収縮に必要なATPというエネルギーの、主要な源であるミトコンドリアはSRと密接に相互接続されており、Ca2 +はこれらのオルガネラの最適な機能に不可欠です。ただし、Ca 2+の蓄積はミトコンドリアの機能を損ない、ATPの生成を減少させ、活性酸素種(ROS)の放出を増加させることがあります。酸化ストレスは心不全(HF)に寄与しますが、ミトコンドリアのCa2 +が心不全においてどのような役割を果たすかは明らかになっていません。ここでは、心筋梗塞HFのマウスモデルにおいて、弛緩時の*2 筋小胞体からの Ca 2+リークがミトコンドリアCa 2+過負荷と機能障害を引き起こすことを、(著者らの知る限りで)初めて示しています。心臓SRには2種類のCa2 +放出チャネルがあります。2型リアノジン受容体(RyR2s)と2型イノシトール1,4,5-三リン酸受容体(IP3R2s)です。 SR Ca2 +リークを引き起こす、または阻害するRyR2変異を含むマウスモデルを使用して、リークのあるRyR2チャンネルがミトコンドリアのCa2 +過負荷、形態異常、および機能不全を引き起こすことがわかりました。対照的に、IP3R2を心臓特異的に欠損させても、HFのミトコンドリアの機能に大きな影響はありませんでした。さらに、ミトコンドリアの抗酸化活性を遺伝学的な手法で強化することにより、ミトコンドリア機能が改善され、RyR2高分子複合体の翻訳後修飾が減少しました。これらのデータは、IP3R2ではなく、漏洩RyR2チャネルがHFでミトコンドリアのCa2 +過負荷と機能障害を引き起こすことを示しています。
HATのヒストンアセチル化が転写開始を引き起こしているという説明が怪しくなってきたところで、さらに大胆な仮説として、ヒストンアセチル化は転写活性化の引き金となっている(primarily cause transcription)のではなく、転写が活性化された跡として残る結果的なもの(consequence of transcription)なのではないか?という問いに挑みます。
今回紹介した論文はbioRxivというプレプリントサーバーに上がっていたものです。日頃からプレプリントにまで目を通しているという人、アラートが来るようにしている人、プレプリント?なにそれ?という人など、色々な方がいると思いますが、”preLights”というサイトをご存知でしょうか?簡単に言うとバイオ系プレプリント論文のまとめサイトで、Journal of Cell Science 誌などを発行するThe Company of Biologistsによって運営されています。
本日は「IP3 Receptors Preferentially Associate with ER-Lysosome Contact Sites and Selectively Deliver Ca2+ to Lysosomes (IP3受容体はER-リソソームの接触部位に集まり、Ca2 +をリソソーム選択的に輸送する)」という論文で、イギリス Department of Pharmacology, University of Cambridge の Dr. Colin W. Taylor のグループによる研究。(論文サイトへのlink→*1)
Inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs) allow extracellular stimuli to redistribute Ca2 + from the ER to cytosol or other organelles.We show, using small interfering RNA (siRNA) and vacuolar H + -ATPase (V-ATPase) inhibitors, that lysosomes sequester Ca2 + released by all IP3R subtypes, but not Ca2 + entering cells through store-operated Ca2 + entry (SOCE) .A low-affinity Ca2 + sensor targeted to lysosomal membranes reports large, local increases in cytosolic [Ca2 +] during IP3- evoked Ca2 + release, but not during SOCE.Most lysosomes associate with endoplasmic reticulum (ER) and dwell at regions populated by IP3R clusters, but IP3Rs do not assemble ER-lysosome contacts. Increasing lysosomal pH does not immediately prevent Ca2 + uptake, but it causes lysosomes to slowly redistribute and enlarge, reduces their association with IP3Rs, and disrupts Ca2 + exchange with ER.In a “piston-like” fashion, ER concentrates cytosolic Ca2 + and delivers it, through large-conductance IP3Rs, to a low-affinity lysosomal uptake system.The involvement of IP3Rs allows extracellular stimuli to regulate Ca2 + exchange between the ER and lysosomes.